 © 2016, AUDRA GERAS/GERAS HEALTHCARE PRODUCTIONS
© 2016, AUDRA GERAS/GERAS HEALTHCARE PRODUCTIONS
Although they are not alive themselves, proteins nonetheless progress through a life cycle of sorts: they are created by the cell, serve a specific purpose in the organism, and ultimately expire either by passive accumulation of structural defects or through active metabolic processes. As in ecological circles of life, the dead are degraded for their core components. But rather than the scavengers and microbial decomposers at work in macroscale habitats, much of the protein recycling work within the cell falls to a barrel-shaped protein complex known as the proteasome.
Found in most known organisms, the proteasome is the crucial component of ubiquitin-mediated protein degradation. It complements the numerous proteases that degrade proteins in the cell. Protease targets can be very broad, even random, yet at the same time, the proteases themselves can be quite limited in the extent to which they break down...
High-resolution structural microscopy techniques and innovative chemical biological approaches have allowed researchers to unveil the different roles proteasomes play in cellular maintenance and adaptation to changing biological environments. Scientists now know there are different kinds of proteasomes, some of which are highly specialized. The constitutive proteasome is present in all cells, while the others—the immunoproteasome, the thymoproteasome, and the spermatoproteasome—are found in immune cells, the thymus, and the testes, respectively.
The proteasome has offered researchers a tool to manipulate levels of a specific protein and test its function, or to treat cancer, inflammation, and other diseases.
The more researchers learn about proteasome structure and function, the better equipped they are to harness its power for their own purposes, turning the cell’s protein decomposer into an invaluable biological apparatus.
Proteasome structure and function
The proteasome weighs in at an impressive 2,500 kilodaltons, making it comparable in size to the ribosome. This beefy complex is composed of more than 60 protein subunits that act together to hydrolyze targeted proteins into short peptides of just 3 to 15 amino acids. These peptides are then broken down further into their constituent amino acids by cellular proteases.
The proteasome can be divided into two main components, the core particle and the regulatory particles. The core particle is formed from four rings, each composed of seven subunits, that are stacked to form the proteasome’s barrel structure, which measures approximately 5 nanometers in diameter—almost as thick as the cell membrane. The two outer rings are constructed of alpha subunits that constrict the ends of the barrel to control access to the lumen. The two inner rings contain proteolytic beta subunits that degrade protein chains as they pass through. The regulatory particles form “lids” on either end of the barrel, unfolding polyubiquitinated target proteins and threading them through the narrowed opening of the core particle into the lumen. The proteasome is a two-way street; proteins can enter and exit either side.
The proteasome is responsible for three types of ATP-dependent proteolytic activity: chymotrypsin-like, which cleaves on the carboxyl side of a target protein’s hydrophobic amino acids; trypsin-like, which chops up the carboxyl side of basic amino acids; and caspase-like, which cuts the carboxyl side of acidic amino acids. Each of these protease activities is encoded in separate beta subunits in the core particle—β5, β2, and β1 subunits, respectively. Despite targeting different parts of a protein for cleavage, all of these subunits act via a similar mechanism: a threonine residue in each β subunit attacks the peptide bond of its target amino acid. Last year, Michael Groll of Technische Universität München and colleagues found that, in order to keep the proteasome from degrading itself during its initial assembly, these reactive threonine residues are masked by protective amino acids that are snipped off in the final assembly step.1
Specialized proteasomes are structurally similar to the constitutive proteasome, but do have some unique alpha and beta subunits that impart functional differences. For example, due to its beta subunits β1i, β2i, and β5i, immunoproteasomes have dramatically reduced caspase-like proteolytic activity and greatly increased chymotrypsin-like activity, which is believed to assist in creating peptide fragments that are better suited for binding to the major histocompatibility complex during antigen presentation.
Conversely, the thymoproteasome-specific β5 subunit (β5t) gives that complex much lower chymotrypsin-like activity than constitutive proteasomes, a difference that appears to be instrumental for the thymus to select CD8+ T cells for survival and maturation. Knockout mice that lack β5t lose more than 80 percent of their cytotoxic CD8+ T cells and succumb to infections that wild-type mice overcome.2 Finally, the altered alpha subunit composition of the spermatoproteasome may contribute to the degradation of acetylated core histones, an important step in chromosome condensation during spermatid differentiation and spermatogenesis.
Proteasome-based medicine
 The structure and function of the cell’s protein-degrading machine
The structure and function of the cell’s protein-degrading machine
See full infographic: WEB | PDF© STEVE GRAEPELThe rapid proliferation of cancer cells demands fast turnover of expired proteins, as well as the efficient disposal of proapoptotic and tumor suppressor proteins. As a result, malignant cells are more susceptible to the cytotoxic effects of inhibiting protein degradation than normal cells.
Over the past couple of decades, scientists have discovered a number of small-molecule inhibitors of the proteasome. Inhibitors belonging to the peptide aldehyde group inhibit the proteasome reversibly and are used extensively in research. However, they can also block the action of cellular proteases, limiting their utility as a research tool and as a potential drug. Beta-lactone proteasome inhibitors bind irreversibly to the active-site threonine of the β subunits in the core particle, but also are not completely proteasome-specific. Members of the boronic acid and epoxyketone classes of inhibitors are more specific for the proteasome and as such have successfully made the leap from research to therapeutic applications.
In 2003, the boronic acid class inhibitor bortezomib (Velcade) became the first therapeutic proteasome inhibitor approved by the US Food and Drug Administration (FDA), which green-lighted the compound as a multiple myeloma treatment. Enormously successful, bortezomib has been shown to have a response rate of 50 percent to 90 percent in multiple myeloma patients who have relapsed following established front-line therapies.
 PROTEASOME ACTIVITY
PROTEASOME ACTIVITY
See full infographic: WEB | PDFTHE SCIENTIST STAFFBortezomib treatments can cause peripheral neuropathy and a decrease in blood platelets, however, thereby limiting the dose that patients can be given. In 2015, the FDA approved an epoxyketone class inhibitor that we helped develop, called carfilzomib (Kyprolis), for the treatment of refractory myeloma. Clinical trials have shown that carfilzomib also has a high patient-response rate, but does not cause the peripheral neuropathy seen with bortezomib.3 Another boronic-acid-class inhibitor, ixazomib (Ninlaro), was approved in 2015 by the FDA for the treatment of myeloma. Unlike carfilzomib and bortezomib, which need to be administered intravenously, ixazomib is formulated to withstand the acidic environment of the stomach and is taken orally.
However, the use of these drugs can lead to the development of resistance. (See “Resist or Desist,” The Scientist, April 2017.) Studies aimed at determining the molecular basis for acquired resistance have found that mutations that confer resistance to bortezomib also reduce carfilzomib effectiveness.4 Thus, the effort to develop the next generation of cancer-fighting proteasome inhibitors continues.
In the meantime, researchers are also exploring the potential of proteasome inhibitors to treat a variety of other diseases. While the inhibitors presented so far block all proteasomes, the specialized biology associated with the nonconstitutive varieties has inspired a hunt for drugs that specifically target a particular type. Given the immunoproteasome’s continuous expression in cells of the immune system, and the stimulation of its production in other tissues during heightened immune activity, researchers reasoned that selective inhibition of the protein-degrading machine might alleviate chronic inflammatory diseases without incurring side effects (neuropathy, platelet suppression) from blocking the other proteasomes. In a mouse model of lupus, inhibition of the immunoproteasome by the compound ONX-0914 halted the progression of the disease state.5 Much research has been conducted to develop inhibitors with even greater selectivity for the immunoproteasome,6 and scientists, clinicians, and patients alike hope that immunoproteasome inhibitors will soon begin clinical testing against autoimmune disorders.
Proteasome inhibition may also be of value in the treatment of neurodegenerative disorders. The process of neurodegeneration is driven in part by inflammation, and inflammation depends on active proteasomes. Indeed, regions of Huntington’s disease–afflicted brains containing the trademark pathological aggregates also possess neurons with high levels of immunoproteasomes that exacerbate the inflamed state of the tissue. Inhibitors would help dampen that neuronal inflammation. In addition, because proteasome inhibitors also increase production of nerve growth factor, which can heal mildly damaged nerve cells, they might actually have a dual-action neuroprotective effect.
Cardiac tissue following oxygen deprivation (ischemia) may also benefit from proteasome inhibition. But, it is critical to limit the inhibitors’ effect to only the chymotrypsin-like activity to protect the heart from damage. Selective inhibition of increased chymotrypsin-like activity during ischemia preserves proteins critical for normal cardiac functioning; but general inhibition of all three proteasome activities can enhance cardiac cell death.
Finally, proteasome inhibitors may prove useful in fighting certain infections. Researchers have begun to develop inhibitors that selectively block the proteasomes in infectious organisms while sparing human proteasomes. An early example is Mycobacterium tuberculosis, an unusual bacterium because it has proteasomes (most bacteria do not).7 Scientists have developed inhibitors that are over 1,000-fold more selective for the M. tuberculosis proteasome over the human counterpart in vitro; the next step is to test their safety and efficacy in people. The emergence of multidrug-resistant strains of M. tuberculosis and the estimated 2 million deaths annually from the disease has led the World Health Organization to declare tuberculosis a global health emergency. Given the urgency, the realization of a clinically useful proteasome inhibitor to combat tuberculosis would be invaluable.
Inhibitors with selectivity for the proteasome found in disease-causing parasites such as Plasmodium falciparum (malaria), Leishmania spp. (leishmaniasis), and Trypanosoma sp. (Chagas disease) are also in varying stages of development. Although not yet sufficiently advanced for clinical testing, these proteasome inhibitors offer a hope to fill the clear and compelling need for new pathogen-fighting agents.
Harnessing the proteasome in basic research
Outside of the clinic, researchers often control the proteasome to manipulate biological systems. Proteasomes are known to play a role in diverse cellular processes, including inflammation, apoptosis, circadian regulation, and more. From the study of any of these phenomena to examining the life cycle of individual proteins, proteasome inhibitors continue to be used in the lab extensively.
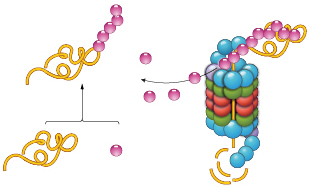
 UBIQUITIN BOOST: PROTACs help catalyze the ubiquitination of target proteins. PROTACs require only brief contact to trigger ubiquitination, meaning that one PROTAC molecule can flag multiple copies of its target for disposal.© STEVE GRAEPEL
UBIQUITIN BOOST: PROTACs help catalyze the ubiquitination of target proteins. PROTACs require only brief contact to trigger ubiquitination, meaning that one PROTAC molecule can flag multiple copies of its target for disposal.© STEVE GRAEPEL
Inhibition, of course, isn’t the only way to manipulate the proteasome. Quite the opposite, in fact. Researchers have devised strategies for directing ordinarily stable proteins to the proteasome for their degradation. While inhibiting the active sites of proteins with a small molecule permits exploration of a single biological activity, degradation of that protein eliminates all of its biological functions and allows for a more thorough consideration of its importance.
An example of such a protein “knockdown” strategy comes from the research group of Tom Wandless at Stanford University. Wandless and his colleagues identified destabilizing peptide sequences that could be spliced into a protein of interest to target it for proteasomal degradation. Wandless also developed a small molecule called Shield-1, capable of binding to and stabilizing the destabilized peptide sequence. Treatment with Shield-1 ultimately prevents recognition of the recombinant protein for ubiquitination and thereby preserves its expression.8 While highly specific as well as adjustable, this application was nevertheless limited since it required the introduction of the recombinant/destabilized protein into the system by genetic modification. This is a relatively simple task in a dish of cultured cells, but at the level of a whole organism, genetic modification is laborious and difficult in mice, let alone humans.
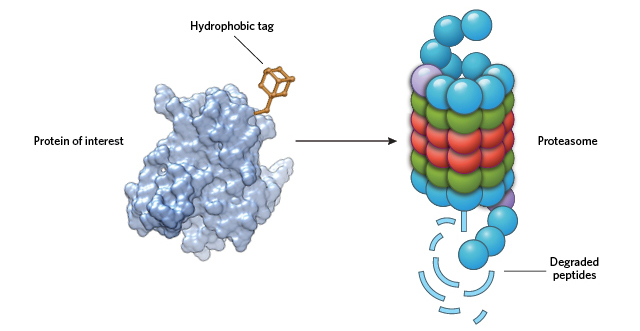 MASQUERADING AS MISFOLDED: By adding a large hydrophobic moiety to a target protein’s surface, HyTs cause the cell to see the protein as unfolded. This activates chaperone proteins in the cell to direct the “unfolded” protein to the proteasome for degradation. © STEVE GRAEPEL
MASQUERADING AS MISFOLDED: By adding a large hydrophobic moiety to a target protein’s surface, HyTs cause the cell to see the protein as unfolded. This activates chaperone proteins in the cell to direct the “unfolded” protein to the proteasome for degradation. © STEVE GRAEPEL
For many years, our lab has been developing strategies to target proteins for posttranslational knockdown without the need for any genetic modification. One class of molecules that promotes protein degradation in such a way is the hydrophobic tags (HyTs), which function by appending a large hydrophobic moiety to the surface of a target protein. The presence of the hydrophobic group mimics a partially unfolded state, thereby engaging the cell’s heat shock proteins and/or other chaperones that maintain protein quality.9 Unable to correct the faux unfolded state of the target protein, the chaperones ferry it to the proteasome for disposal.
This strategy works with many different classes of proteins, including those that lack a tractable active site and therefore cannot be inactivated by a traditional small molecule inhibitor. One compelling target for degradation is the pseudokinase Her3,10 which plays a prominent role in ovarian and breast cancer. Moreover, the use of HyTs to degrade oncogenic proteins could overcome some forms of developed resistance to traditional inhibitor therapeutics. For example, laboratory tests have shown that androgen-based HyTs help fight the growth of castration-resistant prostate cancer cells that can no longer be controlled by androgen receptor antagonists.9
Another class of molecules that causes protein knockdown is known as PROTACs (PRoteolysis TArgeting Chimeras). These two-headed molecules facilitate polyubiquitination of target proteins, thereby tagging them for destruction by the proteasome. As we recently showed, PROTACs catalyze the eradication of specific proteins. One PROTAC molecule causes the degradation of multiple copies of its target.11 PROTACs require only transient contact with their targets to cause their ubiquitination and degradation, as opposed to reversible inhibitors, which need constant occupancy to block target functioning; the latter scenario requires near-saturating concentrations of inhibitor to produce a biological result.
While active site inhibition of proteins with a small molecule permits exploration of a single biological activity, degradation of that protein eliminates all of its biological functions and allows for a more thorough consideration of its importance.
Since the early 2000s, our lab has created PROTACs that effectively cause the degradation of numerous protein targets, including transcription factors, enzymes, scaffolding proteins, and a variety of kinases. Only in the past few years, however, have PROTACs been successfully used in vivo. The earliest PROTACs we constructed included peptide components, which caused them to be quickly degraded in the bloodstream. In 2008, we reported our synthesis of the first PROTAC composed entirely of small molecules,12 and we have since developed a number of others that, owing to their lack of a peptide component, are more metabolically stable and better able to enter cells. For example, our group and others have developed PROTACs that degrade the protein BRD4, which itself drives up levels of the known oncogene, Myc. We have shown that these PROTACs suppress Myc-driven tumors effectively in mouse xenograft models.13,14 With these advances, PROTACs may soon join other proteasome-targeting therapeutics on the path to the clinic.
Most recently, we have designed PROTACs with a greater level of mechanistic sophistication. HaloPROTACs, by virtue of an incorporated chloroalkane group, irreversibly bind their target proteins such that degradation proceeds with greater expedience and with a higher degree of targeting efficiency.15 PhosphoPROTACs make target degradation conditional on the activation of specific signaling pathways, permitting researchers to better map out the connections between downstream effectors and upstream receptors.16
With these and other new techniques, scientists will chip away at several remaining questions about proteasome function. Can we develop effective inhibitors for the prokaryotic proteasome, which is simpler than the proteasome of higher organisms, into an effective target to combat bacterial infections? Are there other tissue-specific proteasomes, like the thymoproteasome and spermatoproteasome, that have yet to be discovered and what are their biological roles? Can small molecules like PROTACs and HyTs be used to target proteins lacking traditional active sites that are currently considered undruggable under the modern pharmaceutical paradigm?
The answers to these questions will serve as the foundation for new and exciting developments that may transform both chemical biological investigations and modern medicine.
Craig M. Crews is a professor at Yale University and the founder of Arvinas, LLC, which focuses on developing protein-degradation therapeutics. John Hines is a research scientist in the Crews lab.
References
- E. Huber et al., “A unified mechanism for proteolysis and autocatalytic activation in the 20S proteasome,” Nat Commun, 7:10900, 2016.
- T. Nitta et al., “Thymoproteasome shapes immunocompetent repertoire of CD8+ T cells,” Immunity, 32:29-40, 2010.
- R. Harvey, “ Incidence and management of adverse events in patients with relapsed and/or refractory multiple myeloma receiving single-agent carfilzomib,” Clinical Pharmacol, 6:87-96, 2014.
- E. Huber et al., “Bortezomib-resistant mutant proteasomes: Structural and biochemical evaluation with carfilzomib and ONX 0914,” Structure, 23:407, 2015.
- H. Ichikawa et al., “Novel proteasome inhibitors have a beneficial effect in murine lupus via the dual inhibition of type I interferon and autoantibody secreting cells,” Arthritis Rheum, 64:493-503, 2012.
- C. Dubiella et al., “Selective inhibition of the immunoproteasome by structure-based targeting of a non-catalytic cysteine,” Angew Chem, 54:15888, 2015.
- G. Lin et al., “Inhibitors selective for mycobacterial versus human proteasomes,” Nature, 461:621-26, 2009.
- L. Banaszynski et al., “A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules,” Cell, 126:995-1004, 2006.
- J. Gustafson et al., “Small molecule mediated degradation of the androgen receptor through hydrophobic tagging,” Angew Chem Int Ed Engl, 54:9659-62, 2015.
- T. Xie et al., “Pharmacological targeting of the pseudokinase Her3,” Nat Chem Biol, 10:1006-12, 2014.
- D. Bondeson et al., “Catalytic in vivo protein knockdown by small-molecule PROTACs,” Nat Chem Biol, 11:611-17, 2015.
- A. Schneekloth et al., “Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics,” Bioorg Med Chem Lett, 18:5904-08, 2008.
- G. Winter et al., “Phthalimide conjugation as a strategy for in vivo target protein degradation,” Science, 348:1376-81, 2015.
- K. Raina et al., “PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer,” PNAS, 113:7124-29, 2016.
- D. Buckley et al., “HaloPROTACS: Use of small molecule PROTACs to Induce degradation of HaloTag fusion proteins” ACS Chem Biol, 10:1831-37, 2015.
- J. Hines et al., “Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs” PNAS, 110:8942-47, 2013.
Interested in reading more?




