
 © MICHELLE KONDRICHReceiving three separate courses of a new class of anticancer immunotherapy agents is not typical for a cancer patient, yet that is what retired Major League Baseball administrator Bill Murray, now 79, endured to treat his melanoma. “When I was told that I might be dying from melanoma, I thought I might as well go for it,” says Murray. In 2011, Murray was given a round of a peptide-based vaccine plus nivolumab (Opdivo), a monoclonal antibody that targets the programmed cell death protein 1 (PD-1) displayed on the surface of T cells, as part of a clinical trial at the Moffitt Cancer Center in Florida. Unfortunately, this two-pronged attack—lasting 12 weeks—didn’t work.
© MICHELLE KONDRICHReceiving three separate courses of a new class of anticancer immunotherapy agents is not typical for a cancer patient, yet that is what retired Major League Baseball administrator Bill Murray, now 79, endured to treat his melanoma. “When I was told that I might be dying from melanoma, I thought I might as well go for it,” says Murray. In 2011, Murray was given a round of a peptide-based vaccine plus nivolumab (Opdivo), a monoclonal antibody that targets the programmed cell death protein 1 (PD-1) displayed on the surface of T cells, as part of a clinical trial at the Moffitt Cancer Center in Florida. Unfortunately, this two-pronged attack—lasting 12 weeks—didn’t work.
PD-1 is a signaling receptor on activated T cells that functions as an immune checkpoint, tamping down T cell activity when it detects its counterpart, PD-L1, on a tumor cell’s surface. Blocking PD-1 was...
Five years later, Murray’s cancer resurfaced. Weber decided to use the most powerful immune checkpoint inhibitor regimen on the market: a combination of nivolumab plus ipilimumab that was approved by the US Food and Drug Administration (FDA) in January 2016. Over the last nine months of 2016, Murray received four doses of the combo, and he will continue to receive nivolumab maintenance therapy for a total of 12 months.
So far, Murray says he is feeling fine, and he even flew from New York to Florida to spend the winter. But he will have to be regularly monitored for cancer over the coming years; there are never any guarantees that tumors won’t return. While immunotherapies provide a better chance for a long-term and durable response, Murray’s story highlights that even this new class of cancer treatments is susceptible to drug resistance, a problem that has plagued the field since the first chemotherapies were used in the United States in the 1940s.1
I’ve been saying this for 15 years: beating cancer takes time, and we need more drugs.—Charles Sawyers
Memorial Sloan Kettering Cancer Center
Just as bacteria evolve resistance to antibiotics, cancer cells evolve ways to evade even the best weapons in medicine’s arsenal. Tumor cells employ numerous tactics—most of which remain unknown—to escape being killed by chemotherapeutic drugs, cytotoxic agents that indiscriminately kill both cancerous and noncancerous cells in the process of dividing. (See “Quest for Chemotherapy Biomarkers” here.) When researchers began to study the genetic mutations (and, eventually, the entire genomes) of tumors, they identified some of the factors required for cancer cell proliferation and survival, including proteins involved in angiogenesis and signaling kinases that, when mutated, fuel tumor growth. This led to the development of drugs that directly bound and disrupted these factors. But even these so-called molecular-targeted therapies were plagued by resistance problems, with rapid tumor shrinkage often followed by regrowth of the cancer weeks or months later.
“I’ve been saying this for 15 years: [beating cancer] takes time, and we need more drugs,” says Charles Sawyers of New York City’s Memorial Sloan Kettering Cancer Center, where he chairs the Human Oncology and Pathogenesis Program.
With the advent of checkpoint-inhibiting antibodies and other powerful immunotherapies, oncologists were optimistic that this new approach to cancer would be less evadable than chemotherapy or molecular-targeted therapies had proved to be. But as Murray’s experience demonstrates, cancer finds a way. Researchers are now zeroing in on the types of resistance that can emerge following checkpoint inhibition, to determine when resistance is likely to arise and to design more-robust therapeutic strategies.
“The big question is whether it’s the tumor cells that are becoming resistant, if the immune system is becoming dysfunctional, or a combination of both,” says Jesse Zaretsky, an MD/PhD student at the University of California, Los Angeles (UCLA) who studies mechanisms of immunotherapy resistance in melanoma.
Countering resistance to targeted therapy
In contrast to the mostly obscure chemotherapy resistance mechanisms, genes and proteins that are likely to change in the evolution of resistance to molecular-targeted therapies are, by and large, predictable. In response, researchers can often devise rational strategies, such as prescribing combinations of drugs that block multiple steps of a tumor growth pathway, to boost a treatment’s chance of successfully eliminating a patient’s cancer.
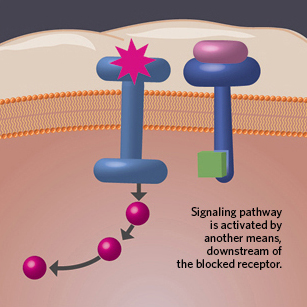 MECHANISMS OF ACQUIRED RESISTANCE: When it comes to molecular-targeted agents and immunotherapies, research has nailed down some basic strategies of cancer's resistance.
MECHANISMS OF ACQUIRED RESISTANCE: When it comes to molecular-targeted agents and immunotherapies, research has nailed down some basic strategies of cancer's resistance.
See full infographic: WEB | PDF© NIRJA DESAIOne of the first examples of nailing down a mechanism of evolved resistance comes from the field of lung cancer. Approximately 10 percent of lung tumors harbor an activating mutation in the epidermal growth factor receptor (EGFR) gene, which encodes a cell-surface receptor that acts as an upstream activator of several pathways (including PI3K and MAP kinase, elements of which are mutated in many cancers). In 2003, the FDA approved the first EGFR inhibitor, gefitinib (Iressa), but within just a few months, some patients stopped responding to the treatment. Two years later, some of the patients whose tumors had become recalcitrant were found to have malignancies that harbored a novel mutation, a methionine-for-threonine substitution at amino acid 790 (T790M) that allowed for continued activation of EGFR despite EGFR inhibitor binding.2,3
“Back then, it was very novel to sequence a gene and find new, acquired mutations and then tell the story of how often these occurred in patients,” says Geoffrey Oxnard, a medical oncologist at the Dana-Farber Cancer Institute in Boston who was involved in identifying mechanisms of acquired resistance to EGFR inhibitors. “It took years and years of collecting and sequencing patient tumor biopsies to understand that biology.” Researchers now know that the vast majority of patients with metastatic EGFR-positive lung cancers eventually develop resistance, and about half those cases are due to a T790M mutation.4
In 2009, researchers developed an EGFR inhibitor that specifically targeted T790M mutation–harboring tumor cells,5 and in 2015, osimertinib (Tagrisso) was approved for clinical use in patients with tumors that are resistant to the first-generation EGFR inhibitors, along with a test that specifically detects T790M mutations. In 2013, the FDA had approved a broader-spectrum tumor tissue test to check for the types of EGFR mutations present. “This is the first example of the FDA saying that a patient needs to have a biopsy to figure out the type of resistance in order to choose the next therapy,” says Oxnard. “What’s cool about this is the potential of science like this going from something discussed in the lab to something that oncologists are using to help treat their patients.” (Last June, the agency approved a blood-based genetic test to detect the same mutations that now allows patients to be tested noninvasively.)
Unfortunately, the fight against cancer’s evolution of resistance is far from over. The same year osimertinib was approved, Oxnard and his colleagues described a mutation, C797S, that rendered patient-derived lung cancer cell lines resistant to the new drug.6 But just as swiftly, the researchers isolated a compound that can bind to this newly identified mutated protein and is effective in killing these tumors in mice.7 “There’s this constant chase of the next resistance mechanism and next therapy,” says Oxnard.
Prostate cancers generally take a different tack in acquiring resistance. Typically treated with drugs that prevent the synthesis or activity of male hormones called androgens, prostate tumors can sometimes restore androgen signaling by activating factors downstream of a drug’s inhibition. In contrast to EGFR-driven lung tumors treated with an EGFR inhibitor, in which resistance mutations arise within the gene encoding the drug’s target, prostate cancer resistance finds a work-around by restoring downstream signaling, including changes in metabolic regulation.8,9
In January 2017, Sawyers and his colleagues identified a third mechanism by which a tumor can bypass a molecular-targeted therapy: a change in cell identity, or “lineage plasticity,” as Sawyers calls it. His lab initially observed that an unexpectedly high proportion of sequenced metastatic prostate tumors that no longer responded to anti-androgen therapy had an inactivating mutation in the tumor suppressor gene, p53. But prior work had shown that a p53 mutation alone does not confer hormone-based resistance, so the lab looked at the sequencing data more carefully and discovered two other mutations—in the RB and PTEN tumor suppressor genes; either of these, in combination with p53 inactivation, resulted in hormone therapy resistance in human cell lines and human prostate tumors engrafted in mice.10 The reason for the resistance turned out to be a shift in cell identity—mutation of RB and p53 led to overexpression of Sox2, which encodes a transcription factor necessary for self-renewal in embryonic stem cells.11 Rather than luminal prostate cells, these doubly mutated cells “are in a multilineage state that does not rely on androgen signaling,” Sawyers says. Lineage plasticity was also previously observed in EGFR-mutated lung cancer patients treated with EGFR inhibitors.12
“One way to look at these resistance mechanisms is to say, ‘This is so depressing,’” Sawyers says. But on a positive note, he adds, if researchers understand how resistance arises, they may be able to overcome it. Two of these resistance mechanisms—the gene expression level changes and subtle lineage switch—are not a result of a genetic change within the tumor, but rather are epigenetic, and are therefore reversible, Sawyers says. By developing ways to modulate protein levels, “in theory we should be able to prevent [these resistance routes] or restore [drug sensitivity].”
Besides clinical and benchtop studies, scientists are turning to in silico methods to understand resistance and ways to combat it. Andrew Read, who studies the evolutionary genetics of disease at Penn State University, recently collaborated on a mathematical model to understand when it is best to use aggressive therapy to try to kill the entire lot of tumor cells or when tumor containment may be best for the patient’s health.13 “The key was to assume that there is competition between [drug-] resistant and sensitive tumor cells,” explains Read. When a tumor rapidly mutates, an attack-all, swift approach may select for the extra-hardy, resistant cells that will take over the entire tumor. In these cases, keeping drug-sensitive, less-aggressive tumor cells in the mix may actually be advantageous for the patient.
 PRIMARY VERSUS ACQUIRED RESISTANCE: Cancer cells can evolve ways to evade a drug’s attack, or they may already be resistant prior to treatment.
PRIMARY VERSUS ACQUIRED RESISTANCE: Cancer cells can evolve ways to evade a drug’s attack, or they may already be resistant prior to treatment.
See full infographic: WEB | PDF© NIRJA DESAI
Picking apart immunotherapy resistance
While immunotherapies are the new kids on the cancer block, Murray and other patients are already forcing researchers to think about the evolution of resistance. Murray appears to have originally had what’s called a “hot” tumor, says Weber—one that has been infiltrated with immune cells and proinflammatory molecules and is thus more likely to respond to a checkpoint inhibitor.14 Murray’s cancer initially retreated after ipilimumab treatment, then likely developed some type of adaptive resistance while retaining enough residual T cells within the tumor to respond to the third round of immune stimulation, Weber says.
The good news is that resistance in patients who initially respond to immunotherapy appears to be less frequent than in patients treated with a targeted therapy, most of whom can be assured that their tumors will eventually become resistant despite treatment. “It’s fair to say that resistance probably occurs less frequently with immunotherapy,” says Walter Urba, an oncologist who specializes in immunotherapy at Providence Health & Services in Portland. But the fact that acquired resistance is infrequent among patients undergoing immunotherapies makes studying the underlying mechanisms difficult. “This makes the science of exploring and understanding resistance a bit of a challenge because, so far, the reports have only had single-digit numbers of patients,” says Matthew Hellmann, an oncologist at the Memorial Sloan Kettering Cancer Center in New York City who specializes in lung cancer and immunotherapy.
To better understand the changes from pre- to posttreatment that may lead to immunotherapy resistance, researchers are beginning to sample immunotherapy-treated tumors in relapsing patients several years after their initial checkpoint inhibitor dose, searching for factors linked with a cancer’s acquired resistance. In a first-of-its-kind study, Antoni Ribas, director of the tumor immunology program at UCLA, and his colleagues combed the whole exomes of metastatic melanoma tumor samples from four patients who had initially responded to the anti-PD-1 antibody pembrolizumab (Keytruda) and then stopped responding, their tumors beginning to grow again months or even years later.
Two of the patients’ posttreatment tumors, but not their pretreatment ones, had loss-of-function mutations in either the Janus kinase 1 or 2 (JAK1 or JAK2) genes, which encode proteins that sense extracellular interferon gamma signaling and convert that into an intracellular response. The mutations likely rendered the tumor cells insensitive to interferon secreted by activated T cells, resulting in less tumor antigen presentation to the immune system and resistance to interferon-induced growth arrest.15 In a third patient, acquired resistance was mapped to a mutation in the gene for the protein beta-2-microglobulin, which is necessary for cells to present major histocompatibility complex (MHC)-linked antigens on their surface; the loss of this gene thus enables tumor cells to hide out and elude recognition by T cells. The researchers did not find any mutations they could mechanistically link to resistance in the fourth patient, “but that’s not to say that someone else won’t find a mechanism that we just didn’t recognize,” says Zaretsky, one of the authors of the work.
“This study, where the authors showed that genetic mutations in key immune processing genes within the tumor can lead to immunotherapy resistance, has triggered the whole field to rethink how tumors adapt to prevent their own destruction by the immune system,” says Ryan Sullivan, a translational researcher who specializes in melanoma at Massachusetts General Hospital.
Another immunotherapy, one that is nearing market approval, is also facing resistance problems: chimeric antigen receptor (CAR) T cells. For these therapies, researchers harvest T cells from a patient’s blood, then modify the T cells, priming them for activation by tumor antigens, multiply the cells in the lab, and infuse them back into the patient. (See “Safety Belts” here.) One such T-cell modification is the addition of an antibody-derived activating receptor to CD19, a protein normally found on the surface of B cells, including those in B-cell malignancies. In the case of T cells modified to target CD19-expressing leukemia cells, the majority of patients will initially respond to therapy, but about 30 percent of responders will soon relapse, seemingly with leukemia cells that no longer carry the surface marker.
But digging into how the cells are able to survive without CD19, which is thought to be required for B-cell growth, Andrei Thomas-Tikhonenko of the University of Pennsylvania discovered the “resistant cells are not actually CD19-negative.” Rather, they express an alternative isoform of the protein that is missing the exon 2–encoded domain, which is where the antibody-derived CAR receptor binds CD19.16 Thomas-Tikhonenko and his colleagues also found that levels of the splicing factor responsible for retaining exon 2 within the protein were lower in the relapsed leukemia cells compared with cells biopsied prior to treatment. The researchers are now trying to reverse this splicing regulation to force exon 2 inclusion, thus rendering the cells susceptible to anti-CD19 CAR T-cell therapies.
Of course, it’s early days for understanding cancer’s evasion of these types of treatments. “The mechanisms of resistance for immunotherapy are literally just being described now,” says Jason Luke, a medical oncologist who conducts melanoma immunotherapy trials at the University of Chicago. For now, researchers continue to monitor the situation.17 Luke is organizing a study in which biopsies of patients’ tumors will be taken throughout their treatment on an anti-PD-1 antibody. Meanwhile, Sullivan at Mass General is sequencing melanoma tumors before and after anti-PD-1 treatment—the same approach used by the UCLA melanoma team. And researchers at Merck, which manufactures the anti-PD-1 antibody pembrolizumab, are also studying the changes that can convert a responding patient into a resistant one.
The big question is whether it’s the tumor cells that are becoming resistant, if the immune system is becoming dysfunctional, or a combination of both.—Jesse Zaretsky
University of California, Los Angeles
Another study, led by Jennifer Wargo of the University of Texas MD Anderson Cancer Center in Houston, aimed to identify biomarkers of response to immune checkpoint inhibitors anti-CTLA-4 and anti-PD-1 antibodies. Response rates to these agents range from 10 to 50 percent, depending on tumor type. Her lab’s recent work showed that the tumor’s genetic makeup, its gene-expression profile, and the tumor microenvironment all influence melanoma patients’ responsiveness to this type of immunotherapy, and that a biopsy early in the course of treatment rather than a pretreatment biopsy was most telling of whether a patient is likely to respond to the treatment.18
And in a follow-up study of tumor whole exome and T-cell receptor sequencing with the same cohort of melanoma patients that first received an anti-CTLA-4 and then an anti-PD1 antibody, Wargo and her team found that a high tumor mutational load and a genome-wide high copy number loss each independently predicted lack of response to the immunotherapies.19 The team also found that if more of the patient’s T cells carried receptors that bound the same target, that individual was more likely to respond to an anti-PD1 but not an anti-CTLA-4 antibody.
“It’s a general theme we and other researchers are learning,” says Wargo. “It’s not a single biomarker that will give us the complete story on response. It’s the combination of the genome, the host immune system, and even environmental factors like the microbiome.”
Identifying mechanisms of immunotherapy resistance will not only help avoid such cases where patients stop responding to treatment, but will likely also shed light on so-called primary resistance, when a cancer never responds in the first place, says David Kaufman, the executive director of New Jersey–based Merck’s oncology translational research division. Indeed, Ribas and his colleagues have already identified some of the same JAK1 or JAK2 mutations found in tumors with acquired resistance to checkpoint inhibitors in tumors of melanoma patients that never responded to this type of immunotherapy.20 Moreover, these tumors did not express PD-L1, indicating that they may use a different checkpoint to disarm the immune system, which accounts for their lack of response to anti-PD-1 antibodies.
“Our finding that some patients can have these mutations before immunotherapy highlights that those tumors had already gone through a process . . . to avoid an immune response,” says Ribas. “Understanding these mechanisms of resistance should allow us to start thinking about [personalizing] immunotherapies, the same way we think about it for targeted therapies for cancer.”
|
Quest for Chemotherapy Biomarkers
“It is striking to see how many studies there have been, and yet there is nothing in clinical practice,” says Ken Olaussen of the Institut de Cancérologie Gustave Roussy in France. “The failure rate is huge.” The lack of reliable biomarkers stems partly from the fact that chemotherapy’s reach is so broad, acting as a nonspecific cytotoxin that damages DNA. Tumor cells employ many tactics to avoid being killed when their DNA is extensively damaged, from altering their metabolism to prevent entry of the drug, to modifying DNA repair pathways and turning off apoptosis. In 2005, following research demonstrating that post-surgery cisplatin chemotherapy improved survival in some lung cancer patients, Olaussen and his colleagues attempted to find a biomarker that correlated with treatment response. They focused on the expression of ERCC1, a gene involved in DNA repair, because such repair pathways are particularly active in tumor cells that are able to resist being killed by cisplatin. Staining patients’ tumor samples with an ERCC1-targeted antibody, Olaussen and his colleagues found that tumors from patients who had minimal levels of ERCC1 protein appeared to derive a greater benefit from the chemotherapy (N Engl J Med, 355:983-91, 2006). “Sensitivity testing to match a cancer with a specific chemo has generally been a tough business. In 2006, this study made us think that we could now characterize lung tumors to decide who should get which chemotherapy,” says medical oncologist Geoffrey Oxnard of the Dana-Farber Cancer Institute in Boston, who was not involved in the research. But attempts to validate ERCC1 protein levels as a biomarker in larger patient cohorts were unsuccessful, because no existing antibody could distinguish between the four different protein isoforms of ERCC1 found in cells, only one of which appeared to be relevant to cisplatin sensitivity in lung cancer patients (N Engl J Med, 368:1101-10, 2013). Olaussen is not giving up. “We showed that the biology is not wrong but that we currently don’t have the tools to measure ERCC1 expression correctly,” he says. “We’re now trying to find new solutions where we combine functional assays or try to develop an antibody that works.” Others have moved on, though. For Oxnard, “the benefit for patients, even if we do find chemotherapy-resistance biomarkers, is likely to be modest.” As a result, chemotherapy studies have been deprioritized in the context of bigger questions about immunotherapy and targeted therapies. “New studies are difficult,” Olaussen says. Few studies include a no-treatment group, which is necessary to deduce changes linked specifically to chemotherapy, he explains, and “there is a general lack of funding to address these questions.” |

References
- V.T. De Vita Jr., E. Chu, “A history of cancer chemotherapy,” Cancer Res, 68: 8643-53, 2008.
- W. Pao et al., “Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain,” PLOS Med, 2:e73, doi:10.1371/journal.pmed.0020073, 2005.
- S. Kobayashi et al., “EGFR mutation and resistance of non–small-cell lung cancer to gefitinib,” N Engl J Med, 352:786-92, 2005.
- G.R. Oxnard et al., “New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer,” Clin Cancer Res, 17:5530-37, 2011.
- W. Zhou et al., “Novel mutant-selective EGFR kinase inhibitors against EGFR T790M,” Nature, 462:1070-74, 2009.
- K.S. Thress et al., “Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M,” Nat Med, 21:560-62, 2015.
- Y. Jia et al., “Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors,” Nature, 534:129-32, 2016.
- V.K. Arora et al., “Glucocorticoid receptor confers resistance to anti-androgens by bypassing androgen receptor blockade,” Cell, 155:1309-22, 2013.
- J. Li et al., “Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer,” eLife, 6:e20183, 2017.
- S.Y. Ku et al., “Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance,” Science, 355:78-83, 2017.
- P. Mu et al., “SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer,” Science, 355:84-88, 2017.
- M.G. Oser et al., “Transformation from non–small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin,” Lancet Oncol, 16:e165-e172, 2015.
- E. Hansen et al., “How to use a chemotherapeutic agent when resistance to it threatens the patient,” PLOS Biol, 15:e 2001110, doi:10.1371/journal.pbio.2001110, 2017.
- P.C. Tumeh et al., “PD-1 blockade induces responses by inhibiting adaptive immune resistance,” Nature, 515:568-71, 2014.
- J.M. Zaretsky et al., “Mutations associated with acquired resistance to PD-1 blockade in melanoma,” N Engl J Med, 375:819-29, 2016.
- E. Sotillo et al., “Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy,” Cancer Discov, 5:1282-95, 2015.
- S. Koyama et al., “Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints,” Nat Commun, 7:10501, doi:10.1038/ncomms10501, 2016.
- P.L. Chen et al., “Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade,” Cancer Discov, 6:827-37, 2016.
- W. Roh et al., “Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance,” Sci Trans Med, doi: 10.1126/scitranslmed.aah3560, 2017.
- D.S. Shin et al., “Primary resistance to PD-1 blockade mediated by JAK1/2 mutations,” Cancer Discov, 7:188-201, doi:10.1158/2159-8290.CD-16-1223, 2017.
Interested in reading more?




