 AUTOIMMUNE TARGETS: This false-color transmission electron micrograph (TEM) displays a slice through a mammalian pancreatic islet cell, the target of an aberrant immune response in type 1 diabetes patients. The red spots in white spaces are membrane-bound secretory granules containing insulin and other hormones to be excreted into the blood. (Nucleus, upper right.)© SPL/SCIENCE SOURCE
AUTOIMMUNE TARGETS: This false-color transmission electron micrograph (TEM) displays a slice through a mammalian pancreatic islet cell, the target of an aberrant immune response in type 1 diabetes patients. The red spots in white spaces are membrane-bound secretory granules containing insulin and other hormones to be excreted into the blood. (Nucleus, upper right.)© SPL/SCIENCE SOURCE
History often repeats itself. More than 100 years ago, one of the world’s leading immunologists, Nobel Laureate Paul Ehrlich, doubted the very existence of autoimmunity, in which the immune system begins to attack healthy tissues. Envisioning a nightmare scenario where the body turns against itself, Ehrlich reasoned that it would be quite improbable. His skepticism regarding this phenomenon, which he termed “horror autotoxicus” (literally, “the horror of self-toxicity”), delayed the acceptance of this concept for another half century—even in the face of compelling clinical examples of immunity gone haywire.
A century of basic research later, scientists...
Most autoimmune disorders are characterized by a focused attack on a particular organ system: the brain and spinal cord become inflamed in multiple sclerosis, the skin is attacked in psoriasis, the joints are pummeled in rheumatoid arthritis, and the intestines are injured in Crohn’s disease and ulcerative colitis. In some cases, autoimmunity strikes only a single cell type in one organ. In type 1 diabetes, the insulin-producing beta cells located in the islets of Langerhans of the pancreas are destroyed or severely injured. This specificity of the autoimmune response is the result of immune reactivity to particular self-antigens. If researchers can identify and target those antigens, they can theoretically develop therapies to strike at the heart of such disorders.
Such a precision approach would overcome a major problem with today’s autoimmune disease treatments, which generally target critical components of our immune systems, making patients susceptible to infections that their own immune responses would normally contain. In a recent Phase 3 clinical trial of a Janus-kinase inhibitor called baricitinib, which works by blocking key biochemical pathways involved in the production of inflammatory molecules, treatment reduced symptoms of rheumatoid arthritis, but patients experienced a dose-dependent two- to fourfold increase in the incidence of shingles compared to placebo.1 Similarly, an antibody to a Velcro-like adhesion molecule called α4 integrin involved in lymphocyte homing to the brain has proven effective in treating relapses in multiple sclerosis. But this approach carries the risk of an often-fatal brain infection known as progressive multifocal leukoencephalopathy (PML). Thus far there have been more than 500 cases of PML in patients taking the α4 integrin antibody, called natalizumab (Tysabri), with an overall rate of about 1 in 250 and the risk increasing with monthly usage beyond a year.2 Therapies that target only those immune cells that are attacking the affected tissue in a particular autoimmune disease would leave the remainder of the immune system, including the portions that fight infection, free to do its job.
This approach, called antigen-specific therapy, has yet to gain traction, however. Part of the problem is that for many autoimmune conditions we simply do not know the identities of the antigens that are recognized and attacked by the immune system. Although researchers have identified the targeted molecules in a few autoimmune diseases, the pharmaceutical industry has been reluctant to invest in antigen-specific therapies. So far no one has succeeded with this approach, and such attempts are thus deemed “nonvalidated” by industry. Lack of support from the pharmaceutical industry, however, has slowed progress toward validating these targeted therapies. Antigen-specific therapy has only rarely been tested in humans, and failures of clinical trials testing antigen-specific therapies in multiple sclerosis, where we do not yet have a convincing understanding of the key antigens driving disease progression, have cast further shadows over our ability to target any autoimmune disease at its roots.
This mind-set has hindered effective drug development for autoimmune diseases compared with therapeutic advances for many other disorders, where the standards of care now include treatments that attack specific mediators of pathogenesis. In the field of allergy, for example, immune desensitization to offending allergens is accepted medical practice. Even in as complex a disease as certain cancers, researchers have successfully targeted the root cause of cells’ prolific growth. Imatinib (Gleevec) is an exemplary drug. It attacks chronic myelogenous leukemia directly at the point of the fundamental mutation that causes the deadly cancer.
Such highly targeted therapeutics can be considered what Ehrlich called “magic bullets.” In autoimmunity we have no such magic bullet. Development of a therapy that turns off the fundamental disease-causing immune pathways languishes behind other approaches that attack critical components of the normal immune response. Indeed, there are no therapies on the market aimed at blocking those immune responses at the core of the disease process, and only a few, for type 1 diabetes, have even progressed to early-stage clinical trials.
Targeting diabetes
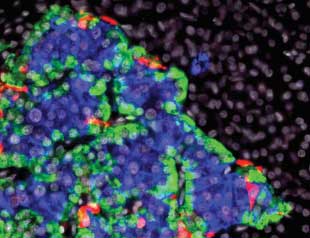 GLOWING ISLET: This composite light micrograph of a pancreatic islet shows beta cells (blue), alpha cells (green), and delta cells (red). © ALVIN TESLER/SCIENCE SOURCEType 1 diabetes is one major autoimmune disease that is ready for a magic-bullet approach. Currently, the only treatment option is insulin, a peptide hormone whose role in maintaining glucose homeostasis was discovered a century ago. Although it has been a lifesaving therapy for type 1 diabetics, insulin does not prevent the long-term, insidious aspects of the disease such as heart attack, stroke, neuropathy, kidney complications, and retinal disease. But the immune responses underlying this widespread disease have been well characterized: both antibodies and cytotoxic “killer” T cells direct attacks on molecules that are produced exclusively in pancreatic beta cells, where the body’s insulin is synthesized. Now, two research teams, including one that I direct, are staging a full frontal assault on these aberrant immune responses, with promising results in early clinical trials.
GLOWING ISLET: This composite light micrograph of a pancreatic islet shows beta cells (blue), alpha cells (green), and delta cells (red). © ALVIN TESLER/SCIENCE SOURCEType 1 diabetes is one major autoimmune disease that is ready for a magic-bullet approach. Currently, the only treatment option is insulin, a peptide hormone whose role in maintaining glucose homeostasis was discovered a century ago. Although it has been a lifesaving therapy for type 1 diabetics, insulin does not prevent the long-term, insidious aspects of the disease such as heart attack, stroke, neuropathy, kidney complications, and retinal disease. But the immune responses underlying this widespread disease have been well characterized: both antibodies and cytotoxic “killer” T cells direct attacks on molecules that are produced exclusively in pancreatic beta cells, where the body’s insulin is synthesized. Now, two research teams, including one that I direct, are staging a full frontal assault on these aberrant immune responses, with promising results in early clinical trials.
One beta-cell antigen (called an autoantigen) targeted in the treatment of type 1 diabetes is proinsulin, the polypeptide precursor of insulin.3 In a 2004 study, researchers followed children from birth and found that the appearance of high-affinity antibodies to proinsulin correlates with the onset of type 1 diabetes. To tolerize patients to their own proinsulin and to quell the inappropriate immune response, my colleagues and I engineered a DNA plasmid encoding proinsulin. The noncoding DNA in the backbone of the plasmid contains naturally occurring hexanucleotide motifs that are immune stimulatory, and are termed CpG sequences. We replaced CpG sequences with a suppressive hexanucleotide sequence, called GpG sequences, that are known to suppress an immune response. An intron was also incorporated into the plasmid to enhance the expression of proinsulin after plasmid injection into muscle.
A precision approach to autoimmunity would overcome a major problem with today’s treatments, which generally make patients susceptible to infections that their own immune responses would normally contain.
Preclinical studies in nonobese diabetic (NOD) mice, which spontaneously develop type 1 diabetes, showed that the engineered plasmid successfully stemmed the autoimmune response and restored normal glucose metabolism.5 The plasmid did so by inducing the expression of proinsulin by antigen-presenting cells (APCs) such as macrophages and muscle cells without the normal costimulation of T cells. To activate T cells and generate an immune response, an antigen like proinsulin must be presented to a T cell by an antigen-presenting cell in the presence of an array of other molecules called costimulatory molecules, such as CD80 and CD86. By expressing proinsulin without costimulation, the engineered plasmid leads to immune tolerance. (See illustration below.) After intramuscular injections of the tolerizing plasmid, the NOD mice experienced an attenuation of the immune response to proinsulin. This resulted in restoration of glucose homeostasis, reduction in islet inflammation, and reduction in antibodies directed against islet cells.
With these preclinical results in hand, we founded a biotechnology company, Tolerion, to sponsor an 80-patient, placebo-controlled clinical trial in the U.S., Australia, and New Zealand in which participants received 12 weekly injections of either the tolerizing plasmid encoding proinsulin or a placebo. The primary endpoint of the trial, which was completed in 2012, was to measure C-peptide, a 31-amino-acid-long section of the proinsulin molecule that is cleaved before secretion from the pancreas as insulin. The level of C-peptide serves as a measure of how well the pancreas is functioning, and C-peptide levels typically decline over time in patients with type 1 diabetes. At the end of the injection period, we observed an increase in the production of C-peptide in the treated individuals, and a drop in C-peptide in the placebo group, compared with C-peptide levels at the start of the trial. We suspect this indicates an improvement in beta cell function in the treated patients. Beta cells that were injured and not yet destroyed may recover function when there is a cease-fire and attenuation of the immune attack. In addition, as C-peptide levels increased, there was a corresponding decrease in cytotoxic T cells that recognized proinsulin.6
Importantly, no T cells that respond to other antigens were affected. This suggests that the plasmid’s effect was antigen specific; tolerance to proinsulin was achieved without altering immunity to viral antigens or to other islet antigens. We are currently planning a follow-up trial in which children with type 1 diabetes will receive weekly dosing of the tolerizing DNA plasmid for one year. Preclinical research has demonstrated that this same plasmid-based approach can provide tolerization to other islet antigens, including glutamic acid decarboxylase, islet-specific glucose-6-phosphatase, and a zinc transporter.5,7 Although the main attack in type 1 diabetes targets proinsulin, these other islet cell molecules may also play a role in the disease, and they too can be targeted.
 TARGETING TYPE 1 DIABETES: In patients with type 1 diabetes, the immune system generates antibodies and activates cytotoxic “killer” T cells to attack molecules that are produced by the beta cells of the pancreas. Researchers are now developing therapies that quell these specific immune pathways to tolerize patients to their own autoantigens while leaving the rest of the immune system intact.
TARGETING TYPE 1 DIABETES: In patients with type 1 diabetes, the immune system generates antibodies and activates cytotoxic “killer” T cells to attack molecules that are produced by the beta cells of the pancreas. Researchers are now developing therapies that quell these specific immune pathways to tolerize patients to their own autoantigens while leaving the rest of the immune system intact.
See full infographic: WEB | PDF© SCOTT LEIGHTONAnother exciting autoimmunity treatment strategy aims at inducing regulatory cells that might suppress unwanted immune activity. First posited by Ehrlich as one way the body prevents autoimmunity from wreaking havoc, regulatory T cells (Tregs) are now known to play an important role in winding down immune responses. Ramping up the production of such regulatory immune cells could provide another avenue for quelling the autoimmune response of type 1 diabetes.
In our preclinical experiments with the proinsulin plasmid, for example, the NOD mice began to produce a regulatory T cell that secreted the immune-suppressive cytokine known as interleukin-10 when stimulated with proinsulin. To harness such natural immune control, Jeffrey Bluestone of the University of California, San Francisco, and colleagues extracted Tregs from 14 type 1 diabetics, expanded the cells in the lab, then reinfused the cells back into the patients. There were no infusion reactions or significant adverse effects, and some of the recipients experienced a slowed decline in their C-peptide levels that lasted up to two years following the treatment.8
Other therapies in clinical trials for type 1 diabetes aim to increase the number of Tregs in the pancreas. But so far, all of these attempts have focused on Tregs that are not generated in response to a specific antigen. Thus, like existing treatments for autoimmune disorders, such therapies might increase the risk of opportunistic infection by affecting a broad swathe of immune cells. Researchers are now working to refine this approach to target a regulatory cell governing only responses to the pancreatic beta cell.
Earlier this year, for example, the University of Calgary’s Pere Santamaria and colleagues tested such a targeted therapy in a variety of mouse models of autoimmunity. The researchers used engineered nanoparticles coated with relevant autoimmune peptides and portions of the major histocompatibility complex, which normally help activate cytotoxic T cells. Without additional costimulatory signals, these nanoparticles instead triggered the in vivo differentiation of self-reactive T cells into antigen-specific Tregs. In animal models, the nanoparticles have resolved diverse autoimmune phenotypes, including diabetes.9 If these results hold up in humans, therapies that aim to boost levels of relevant Tregs could serve as another type of antigen-specific approach to quelling the aberrant immune responses that plague patients with type 1 diabetes.
Treating other autoimmune diseases
Such progress in type 1 diabetes gives hope that an antigen-specific approach might succeed in treating other autoimmune diseases for which the underlying immune pathways are known. And, thanks to many years of bench work and advances in our understanding of human immunity, there are now a handful of disorders for which these immune details are coming to light.
In the vast majority of individuals with myasthenia gravis, for example, the immune system generates antibodies that attack the receptor for acetylcholine on the muscle side of the synapse between motor neurons and muscle cells. Such antibodies are highly pathogenic; a pregnant mother with myasthenia gravis can deliver a myasthenic newborn due to the transfer of such anti-acetylcholine receptor antibodies across the placenta. Presenting the body with acetylcholine receptor (AChR) without the necessary costimulatory factors to trigger an immune response, then, may help tolerize the immune system to this autoantigen, and reduce or stop the production of the disease-causing antibodies against acetylcholine receptors. Indeed, injection of an AChR-encoding plasmid, developed by Tolerion, reduced disease severity in an animal model of myasthenia. Other approaches involving nasal application of peptide fragments of AChR have also shown efficacy in animal models.10 Despite such promising results, no therapies have yet entered clinical trials.
There have been a few trials of antigen-specific therapy in multiple sclerosis (MS). Specifically, my group and others have designed therapies to tolerize MS patients to various proteins of the myelin sheath that surrounds nerve axons in the brain and spinal cord and is attacked by the immune system. However, we simply do not know which of the dozen myelin proteins and several dozen lipids are the targets of the autoimmune affront.11 Perhaps it should not be surprising that early clinical trials have yielded disappointing results. In a 267-patient, Phase 2 trial of a plasmid encoding myelin basic protein—the major protein of the brain’s white matter—the reduction in brain lesions was much less than that seen with the already approved drugs for MS that quell the immune system more broadly.12 Clearly, if one is to succeed with antigen-specific tolerance to treat autoimmune disease, one must have a solid understanding of the antigenic targets involved in the pathogenesis of the disease.
Unfortunately, these recent failures have likely added to prolonging the pharmaceutical industry’s resistance to the development of antigen-specific treatments for autoimmunity. Once again, we may take a lesson from history. Resistance to the development of Gleevec in the 1990s was fierce, and progress was slow, delaying trials for approximately five years. The drug’s developer, Ciba Geigy (now Novartis), was initially reluctant to advance the drug to the clinic, with concerns ranging from Gleevec’s potential toxicities to the small size of the market. These days, similar worries plague the pharmaceutical industry when presented with developing antigen-specific therapies for autoimmune diseases.
With Gleevec, its academic developers Brian Druker and Charles Sawyers fought this resistance and convinced a reluctant pharmaceutical company to proceed. Results in the clinic were stunning, with chronic myelogenous leukemia patients surviving and experiencing long-term remissions of a previously fatal disease. Once the key antigen or antigens that are the targets of autoimmunity in a particular disease have been discovered, scientists should take a similar chance on developing antigen-specific therapies for those diseases. Perhaps then they would achieve similarly stunning results.
Lawrence Steinman is a professor of pediatrics and neurological sciences at Stanford University. He is a founder and member of the board of directors of Tolerion, and serves on the board of directors for Atreca and the scientific advisory boards for Transparency Life Sciences, Receptos (Celgene), TG Therapeutics, Raptor, and Teva.
References
- M.C. Genovese et al., “Baricitinib in patients with refractory rheumatoid arthritis,” New Engl J Med, 374:1243-52, 2016.
- C. Warnke et al., “PML: The dark side of immunotherapy in multiple sclerosis,” Trends Pharmacol Sci, 36:799-801, 2015.
- G.S. Eisenbarth, J. Jeffery, “The natural history of type 1 diabetes,” Arq Bras Endocrinol Metabol, 52:146-55, 2008.
- P. Aschenbach et al., “Mature high-affinity immune responses to (pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes,” J Clin Invest, 114:589-97, 2004.
- N. Solvason et al., “Improved efficacy of a tolerizing DNA vaccine for reversal of hyperglycemia through enhancement of gene expression and localization to intracellular sites,” J Immunol, 181:8298-307, 2008.
- B.O. Roep et al., “Plasmid encoded proinsulin preserves C-peptide while specifically reducing proinsulin specific CD8 T cells in type 1 diabetes,” Sci Transl Med, 5:191ra82, 2013.
- P. Gottlieb et al., “Clinical optimization of antigen specific modulation of type 1 diabetes with the plasmid DNA platform,” Clin Immunol, 149:297-306, 2013.
- J.A. Bluestone et al, “Type 1 diabetes immunotherapy using polyclonal regulatory T cells,” Sci Transl Med, 315:315ra189, 2015.
- X. Clemente-Casares et al., “Expanding antigen-specific regulatory networks to treat autoimmunity,” Nature, 530:434-40, 2016.
- D. Barchan et al., “Antigen-specific modulation of experimental myasthenia gravis: Nasal tolerization with recombinant fragments of the human acetylcholine receptor subunit,” PNAS, 96:8086-91, 1999.
- L. Steinman, “The re-emergence of antigen-specific tolerance as a potential therapy for MS,” Mult Scler, 21:1223-38, 2015.
- H. Garren et al., “Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis,” Ann Neurol, 63:611-20, 2008.
Interested in reading more?




